Einleitung
Eine Lungenfibrose ist eine Krankheit, die zu einer pathologischen Bildung von Narbengewebe in der Lunge führt, sodass diese in ihrer Funktion, den Körper mit Sauerstoff zu versorgen, zunehmend eingeschränkt wird. Dieser Zustand verschlechtert die Lebensqualität des betreffenden Patienten in erheblichem Maße. In einigen Fällen kann das Fortschreiten der Erkrankung zu Lungen- und/ oder Herzversagen sowie im schlimmsten Falle zum Tod führen. Moderne Medikamente können die pathologischen Veränderungen der Lunge nicht rückgängig machen. Alternative Methoden, diese Krankheit zu behandeln, können den Einsatz von Stammzellen umfassen, da ihre erforschten Eigenschaften die Fähigkeit beinhalten, den Entzündungsprozess aufzuhalten sowie die Erholung des geschädigten Gewebes zu stimulieren.
Über Lungenfibrose
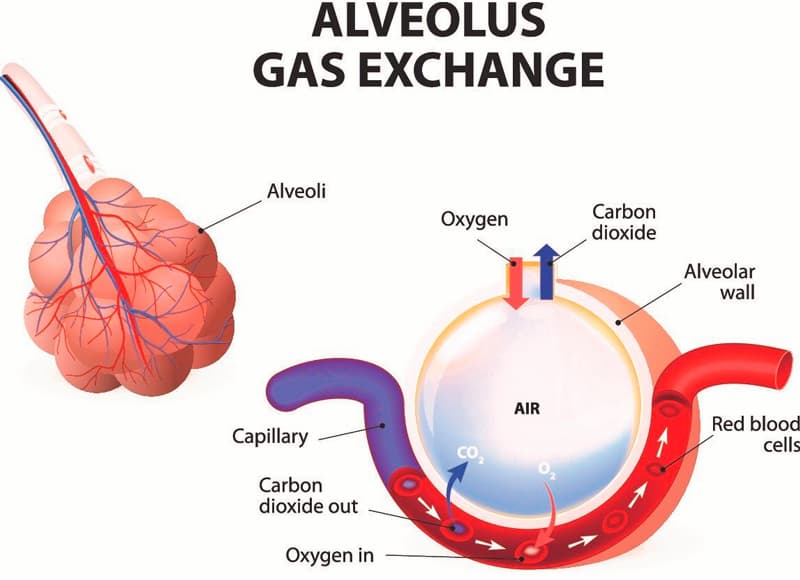
Eine Lungenfibrose ist mit der Vernarbung des Lungengewebes durch fibrotische Veränderungen assoziiert. Das Wachstum des Narbengewebes wird durch eine Entzündung verursacht und führt zu einer Kompression der Alveoli, die nicht länger ihre Funktion erfüllen können, wodurch der Gasaustausch im Körper gefährdet ist.
Alveoli sind die Lungenvesikel, in denen eingeatmete Luft in Kontakt mit dem Blut kommt. Läsionen der Fasern können einen Teil der Lunge oder das gesamte Organ in einem oder beiden Lungenflügeln beeinträchtigen.
Eine Lungenfibrose ist eine progressive Krankheit, die irreversible Schädigungen der Lungengewebe verursacht, deren Komplikationen und Konsequenzen die Folgenden umfassen:
- Chronisches Atemversagen (Sauerstoffinsuffizienz im Körper).
- Lungenhochdruck (hoher Blutdruck in den Lungenarterien).
- Chronische Lungen- Herz und andere kardiovaskuläre Krankheiten [1].
- Lungenkrebs [2].
- Sekundäre Atemwegsinfektionen (einschließlich der Entwicklung einer Pneumonie mit hohem Sterberisiko) [3].
Die Gründe für eine Lungenfibrose
Es ist nicht immer ersichtlich, was genau die Vernarbung der Lunge verursacht. Ein pathologisches Wachstum des Lungengewebes kann durch Allergien, Infektionen der Lunge, genetische Faktoren, Strahlung, Traumata und anderen Prozesse und Umstände ausgelöst werden.
Wenn der genaue Grund der Erkrankung nicht erkennbar ist, sprechen wir von einer idiopathischen Lungenfibrose (IPF). Bei einer solchen Erkrankung wird eine Kombination aus Umweltfaktoren, genetischen und anderen nicht identifizierten Faktoren als Auslöser der Krankheit angesehen.
Es gibt ebenfalls Hinweise, dass Rauchen (sowohl in der Gegenwart als auch in der Vergangenheit) sowie eine belastete Familienhistorie (bei bis zu 20% der Patienten beobachtet) eine Rolle spielen [4]. Wenn mindestens zwei primäre biologische Familienmitglieder (Elternteil, Geschwisterkind oder Kind) eine klinische Manifestation einer IPF hatten, wird die Erkrankung als familiäre Pneumonie (FIP) klassifiziert [5]. Menschen, die ersten Grades mit Patienten mit FIP verwandt sind haben ein stark erhöhtes Risiko, eine Interstitielle Lungenerkrankung (ILD) zu entwickeln.
Interstitielle Lungenfibrose
Eine Interstitielle Lungenfibrose (ILD), auch bekannt als diffuse parenchymale Lungenerkrankung (DPLD), ist eine Gruppe von Erkrankungen, einschließlich Lungenfibrose, bei denen die interstitiellen Gewebe (Bindegewebe der Lunge) geschädigt sind. ILD wird mit einer Entzündung und Vernarbung des Lungengewebes assoziiert.
Interstitielles Gewebe ist das Bindegewebe der Lungen, das als Rückgrat für die Alveoli und Blutgefäße dient. Bei einer Fibrose werden aufgrund des exzessiven Wachstums des Bindegewebes die Alveoli komprimiert und nach und nach durch Bindegewebe ersetzt. Dies resultiert in einem erschwerten Gasaustausch in den Alveoli, was wiederum in einer Hypoxie mündet – der Körper beginnt unter dem Sauerstoffmangel zu leiden.
Ungleich einer Idiopathischen Lungenfibrose (IPF), kann der Grund für eine ILD zuverlässig bestimmt werden. Mögliche Gründe umfassen:
- infektiöse Läsionen;
- autoimmune Erkrankungen/ Erkrankungen des Bindegewebe;
- genetische Prädisposition [6];
- bösartige Tumore;
- das Inhalieren schädlicher Substanzen (Quecksilber-Dampf, Mineralstaub etc.);
- bestimmte Medikamente (beispielsweise einige antiarrhythmische Drogen, Nitrofurantoin und andere) [7].
Kontaktieren Sie uns
Erhalten Sie eine kostenfreie online-Beratung und erfahren Sie mehr über die zu erwartenden Resultate einer Stammzelltherapie >>>

Medizinische Beraterin, Ärztin bei Swiss Medica
Fibrose infolge einer Erkrankung an COVID-19
Die Coronavirus-Pandemie hat die Anzahl aller Lungenfibrose-Erkrankungen erhöht. Es ist bekannt, dass die Vernarbung der Lunge eine der häufigsten und ernsthaftesten Konsequenzen einer Virusinfektion des menschlichen Körpers ist [8]. Der Mechanismus der Entwicklung einer Lungenfibrose infolge einer COVID-19-Erkrankung ist mit einer inkorrekten Immunantwort assoziiert, die zu einer Schädigung des Lungenepithels und der mikrovaskulären endothelialen Zellen führt; insbesondere in Kombination mit einer künstlichen Lungen-Ventilation.

Zudem wird aufgrund von Forschungsberichten und statistischen Daten immer deutlicher, dass Patienten mit milden COVID-19-Infektionen auch nach einer Besserung und Entlassung aus dem Krankenhaus ein verstecktes potentielles Risiko haben, eine Lungenfibrose zu entwickeln [8].
Um die Entwicklung einer Lungenfibrose infolge einer Coronaerkrankung zu verhindern, ist es wichtig, die Behandlung dieser Komplikation so früh wie möglich zu beginnen.
Anzeichen & Symptome einer Lungenfibrose
Der klinische Grund für eine Lungenfibrose ist assoziiert mit einer Abnahme des Lungenvolumens, das den Körper dazu nötigt, sich an diese negativen Konsequenzen anzupassen. Kompensatorische Veränderungen manifestieren sich in einer Atemknappheit – das am meisten verbreitete initiale Symptom einer IPF. Zuerst tritt eine Atemnot nur während physisch anstrengender Tätigkeiten auf; später jedoch auch im Ruhezustand.
Ein Patient mit IPF ist auch mit anderen Symptomen wie trockenem Husten, chronischer Müdigkeit und unerklärlichem Gewichtsverlust konfrontiert.
Klinische Merkmale einer zugrundeliegende Bindegewebserkrankung (CTD) wie zum Beispiel Arthralgie (Gelenkschmerzen) oder Sicca-Symptome (Trockenheit der exokrinen Drüsen, insbesondere der Augen und des Mundes) können ebenfalls beobachtet werden.
Das häufigste äußere Merkmal von IPF ist als “keulenförmige Finger” bekannt; einer Deformation der Finger und Zehen charakterisiert durch die Vergrößerung der Spitzen der Gliedmaßen. Dieses Symptom ist assoziiert mit einem chronisch niedrigen Sauerstoffgehalt des Blutes [9].
Das initiale Stadium der Krankheit geschieht häufig symptomfrei. Zum Beispiel können bei asymptomatischen Individuen mit Risiko für eine familiäre Bindegewebspneumonie (FIP) Anzeichen geweblicher Veränderungen der Lungengewebe diagnostisch nachgewiesen werden [10].
Diagnostik einer Lungenfibrose
Es gibt die Ansicht, dass Lungenfibrosen nicht ausreichend diagnostiziert werden [11]. Meistens wird eine solche Erkrankung erst in einem recht späten Stadium erkannt. Meist wird sie durch eine Abnormalität in einer hochauflösenden Computertomographie (HRCT) der Brust bemerkt [12].
Es gibt Beweise dafür, dass eine Brust-Radiographie bei der Evaluation von Patienten mit vermuteter IPF weniger nützlich ist als eine HRCT [9].
Bronchoskopien mit einer bronchoalveolären Spülung (BAL) und einer transbronchialen Biopsie werden ebenfalls genutzt, um den Zustand der Lungengewebe zu untersuchen.
Des weiteren können laboratorische Diagnosen verschiedener Indikatoren (biologischer Marker) objektiv messbare Daten über sowohl normale als auch pathologische Prozesse des Körpers liefern. Bei einer Lungenfibrose helfen diese Biomarker, die Erkrankung zu diagnostizieren, die Suszeptibilität der Erkrankung offenzulegen und die Wirksamkeit spezifischer Medikamente vorauszusagen [9]. Sie können auch akkurate Aussagen zum Grund des Entstehens der Erkrankung und Prognosen für Patienten mit IPF ermöglichen [13].
Traditionelle und moderne Behandlungen von ILD & Lungenfibrosen
Während Lungenfibrosen viele Jahre brauchen, um sich zu entwickeln, wird diese Erkrankung bei den meisten Patienten erst in einem fortgeschrittenen Stadium erkannt, wenn nicht mehr viel getan werden kann, um ein Fortschreiten der Krankheit zu verhindern. Die bereits existierenden Behandlungen zielen auf die Erhaltung der Lungenfunktionen des Patienten und einen akzeptablen Lebensstandart.
Bei den verbreiteten Behandlungsmethoden einer Lungenfibrose werden adrenale Kortex Hormone (Prednison) benutzt. sind diese nicht effektiv genug, werden Azathioprine un Cyclophosphane eingesetzt. Des weiteren wird Sauerstoff zur Inhalation zugeteilt. Aktuell wurde gezeigt, dass die Medikamente Pirfenidone und Nintedanib die Entwicklung von IPF verlangsamen können [14].
Doch eine Lungenfibrose bleibt eine progressive Krankheit und es sind keine antifibrotischen Mittel oder Behandlungen (abgesehen von einer Lungentransplantation) bekannt, die die schlechte Überlebensrate dieser Krankheit beeinflussen könnten [15].
Alternative Methoden einer Behandlung einer Lungenfibrose schließen den Einsatz von Stammzellprodukten ein. Pathologische Veränderungen einer fibrotischen Lunge werden als irreversibel angesehen, da der Körper nur über sehr schwache Möglichkeiten verfügt, Narbengewebe zu heilen. Doch wenn Stammzellen in der erforderlichen therapeutischen Dosierung in den Körper injiziert werden, ist dies häufig der Beginn des Regenerationsprozesses und verlansamt den Vernarbungsprozess des Lungengewebes [16]. Diese Zellen werden entweder durch einen IV-Tropf (intravenös) oder durch Inhalation für eine gezielte Lieferung der Zellen in die geschädigten Bereiche verabreicht.
Kontaktieren Sie uns
Kontaktieren Sie unseren medizinischen Berater, um zu erfahren, welche Vorteile Ihnen eine Stammzelltherapie bieten kann >>>
Medizinische Beraterin, Ärztin bei Swiss Medica
Stammzelltherapie für Lungenfibrosen
Eine zellbasierte Therapie ist bekannt als eine moderne experimentelle Behandlung, die die körpereigenen Fähigkeiten, sich selbst zu heilen, nutzt.
Wie wirken Stammzellen bei einer Fibrose? Die Besonderheit der “Arbeit” der Stammzellen im Körper liegt darin, dass sie auf Signale von Zellen geschädigter Gewebe reagieren, was die Bildung neuer benötigter Zellen stimuliert.
Studien mit allogenischen (Spender) mesenchymalen Stammzellen (MSCs) haben gezeigt, dass sich in geschädigten Bereichen sammeln und zu der Regeneration der Gewebe beitragen, die chronische Entzündung der Atemwege lindern und das Flüssigkeitsgleichgewicht der Alveolen bei akuten Lungenverletzungen wiederherstellen [17].
Derzeit werden weitere klinische Versuche mit Stammzellen für die Behandlung von Lungenerkrankungen durchgeführt [18].
Was sind mögliche Ergebnisse einer Stammzelltherapie?
Beim Einsatz von Stammzellen für intravenöse Infusionen bei der Behandlung von Lungenfibrosen konnten die folgenden Ergebnisse erzielt werden:
- Die Verlangsamung oder das Beenden des Vernarbungsprozesses durch die “Beruhigung” des Immunsystems des Patienten (Stammzellen haben eine entzündungshemmende Wirkungen).
- Partielle Regeneration des Lungengewebes.
- Bildung eines neuen vaskulären Netzwerkes, das die Erneuerung des Gewebes befördert.
- Linderung der Symptome des Patienten.
Laut einer klinischen Untersuchung steigert die endobronchiale Infusion von Stammzellen die Lebensqualität erheblich. 86% der Studienteilnehmer, die über 12 Monate hinweg untersucht wurden, zeigten einen stabilen funktionalen Status und eine stabile physische Leistung [19].
Die Hauptaufgabe einer stammzellbasierten Therapie ist es daher, das Fortschreiten der Krankheit zu verlangsamen, die Lungenfunktionen zu stabilisieren und den aktuellen Zustand des Patienten beizubehalten.
Sehen Sie eine Video-Rückschau von Patienten mit COPD (auf Italienisch) und erfahren Sie, wie eine Stammzellentherapie bei einer Erkrankung der Lunge helfen kann:

Ein weiterer Rückschau über die Behandlung von Stammzellen bei Lungenfibrose:

Sicherheit der Stammzelltherapie bei einer Lungenfibrose
Klinische Untersuchungen von Therapien basierend auf adulten Stammzellen zeigten ein akzeptables Sicherheitsprofil dieses Ansatzes. Insbesondere wurde berichtet, dass während der gesamten Periode einer Studie mit 72 Infusionen keinerlei Nebenwirkungen beobachtet wurden, die ernsthaft und klinisch signifikant sind [19].
Derzeit wird aufgrund der gemeinsamen Herkunft diskutiert, ob sich MSCs in Fibroblasten differenzieren können und damit die fibröse Kaskade beschleunigen könnten. Es gibt jedoch weder präklinische oder Humanstudien, die diese Beziehung belegen [19].
Bei keinem der Patienten entwickelte sich eine abnormale Gewebeformation. Dies ergab ein Ganzkörper-CT-Scan der 12 Monate nach der ersten Verabreichung von Stammzellen. Das gleiche positive Sicherheitsergebnis konnte 24 Monate nach der ersten Infusion [17] festgestellt werden.
Eine weitere Studie zeigte, dass Infusionen von Stammzellen von allen Patienten gut toleriert wurden. Es traten keinerlei ernsthafte schädliche Vorkommnisse in Folge der Behandlung auf [19].
Kontaktieren Sie uns
Kontaktieren Sie unseren medizinischen Berater und erhalten Sie eine kostenfreie online-Evaluation Ihrer persönlichen Krankheitsgeschichte >>>
Medizinische Beraterin, Ärztin bei Swiss Medica
Quellenangaben:
World Health Organisation. The top 10 causes of death. 24 May 2018.
Mesenchymal Stem Cell as Therapeutic Modality in Interstitial Pulmonary Fibrosis. Clinical Trial.
MD, Pediatrician, Regenerative Medicine Specialist